Home / News
Congratulations! Cheng's work on assessing completeness and contamination of predicted viral contigs has been accepted by ECCB.May 28, 2026 Viruses represent the most abundant biological entities on Earth, playing vital roles in diverse ecosystems. Cataloging viruses across various environments is essential for understanding their properties and functions. Metagenomic sequencing has emerged as the most comprehensive method for virus discovery. However, distinguishing viral sequences from the vast background of microbial organisms in metagenomic data remains a significant challenge. Existing tools experience varying degrees of false positive rates due to noise in sequencing and assembly, and the integration of proviruses into microbial genomes. This highlights the urgent need for an accurate and efficient method to evaluate the quality of viral contigs. To address these challenges, we introduce ViralQC, a tool designed to assess the quality of viral contigs or bins. ViralQC identifies microbial contamination within putative viral sequences using an ensemble framework powered by DNA and protein foundation models and estimates completeness by analyzing protein organization. We evaluated ViralQC on multiple datasets and compared its performance against the state-of-the-art tool, CheckV. Leveraging both DNA and protein foundation models, ViralQC achieves higher sensitivity on contamination detection for contigs longer than 10 kbp while maintaining comparable accuracy. Additionally, ViralQC delivers more accurate estimation on contigs with completeness>50%. The tool is available at Github |
Congratulations! Fuchuan's work on giant virus host prediction has been accepted by ECCB.May 28, 2026 Nucleocytoplasmic large DNA viruses (NCLDVs) play crucial roles in global ecosystems. Although metagenomics has vastly accelerated the discovery of novel NCLDVs, predicting their hosts from fragmented contigs remains a critical bottleneck, with no dedicated end-to-end computational tools currently available. Addressing this gap requires overcoming three fundamental challenges: the extreme scarcity of labeled reference genomes, the severe domain shift between laboratory isolates and diverse environmental metagenomes, and the inability of traditional deterministic models to quantify prediction uncertainty—a crucial requirement for reliable ecological profiling where novel, divergent viruses are prevalent. We present GiantHost, the first NCLDV host prediction tool with domain adaptation and uncertainty awareness. GiantHost employs a dual-tower neural network to integrate dense genome traits and sparse GVOG profiles, allowing better integration of heterogeneous features. To overcome label scarcity and domain shift, we leverage 1,400 environmental viral genomes (GVMAGs) via semi-supervised multi-task learning and Domain Adversarial Neural Networks (DANN), effectively bridging the distributional gap between RefSeq and environmental data. Additionally, GiantHost incorporates Conformal Prediction (CP) to output statistically guaranteed prediction sets rather than overconfident single labels. Evaluated under rigorous genome-level cross-validation, GiantHost demonstrates robust predictive power. Applied to the Tara Ocean dataset, GiantHost successfully captured the vertical stratification of NCLDV hosts—revealing a depth-dependent decline of phytoplankton-infecting viruses and a relative enrichment of Amoebozoa-infecting viruses in the mesopelagic zone. The tool is available at Github |
Congratulations! Yi's work on DNA inversion events detection has been accepted by ECCB.May 28, 2026 Bacterial phase variation enables reversible phenotypic switching and is often mediated by DNA inversion events known as invertons. Although metagenomic sequencing provides a powerful way to study invertons without cultivation, detecting these events remains challenging when sequencing depth is low. In this work, we developed TPMM, a three-component posterior mixture model for probabilistic inverton calling from metagenomic data. TPMM incorporates sequencing depth and read evidence to classify candidates flanked by inverted repeats into noise, low-probability, or high-probability inversion signals. It further assigns posterior probabilities as soft labels and uses cumulative Bayesian False Discovery Rate control to identify reliable invertons. On two real gut metagenomic datasets, TPMM showed strong agreement with PhaseFinder at high depth and recovered substantially more invertons under downsampling, demonstrating improved performance in sparse-data settings. Additional analyses of viral genomes suggest that inversion-mediated regulation may have a broader biological scope. The tool is available at Github |
Congratulations! Dehan's work on viral reassortment identification for segmented viruses has been accepted for publication in Nucleic Acids Research.Mar. 12, 2026 Segmented viruses, including influenza A viruses, can generate novel strains through reassortment, a process in which genome segments from different strains are exchanged. Because reassortment can lead to important changes in viral properties such as transmission, efficient detection of emerging reassortant strains is essential. In this work, Dehan developed VReassort, a fast and accurate tool that identifies reassortment events from genome sequences using deep learning and phylogenetic tree-derived features. VReassort achieved strong performance on both simulated and real datasets, with F1-scores above 0.8, and analyzed about 1,000 influenza A virus strains in under two minutes, making it more than 100 times faster than benchmark methods. Applications to large-scale influenza A and rotavirus datasets further demonstrated its effectiveness and broad potential. The paper will be available soon at Here, and the tool is available at Github |
Congratulations! Yongxin and Jiaojiao's work on plasmid characterization and retrieval has been published on Genome Biology.February 07, 2026 Plasmids play a pivotal role in the emergence of multidrug-resistant and pathogenic bacteria, posing significant clinical challenges. However, the rapidly growing number of unannotated plasmids necessitates comprehensive characterization of their diverse properties. Here, we present PlasRAG, a tool that integrates multi-faceted property characterization of query plasmids and plasmid DNA retrieval based on textual queries. PlasRAG employs a bidirectional multi-modal information retrieval model that aligns DNA sequences with textual data, effectively overcoming the limitations of traditional approaches. Rigorous experiments demonstrate that PlasRAG delivers robust performance and enhanced analytical capabilities, underscoring the effectiveness of its architectural design. The paper is available on Here. |
USTC Professors and Students Visit for Academic Discussion!Dec. 4, 2025 We were delighted to host Prof. Hua Zhengshuang, Prof. Cai, Prof. Mu, and their students from the University of Science and Technology of China (USTC) on Thursday! The visit included engaging discussions about archaea, and we extend our heartfelt thanks to Prof. Hua for the inspiring talk. It was a pleasure to connect and share ideas—looking forward to future collaborations! |

|
Congratulations to Jiayu! Phage-Host Interaction Work was Published in Briefings in Bioinformatics!Nov. 24, 2025 We are excited to share that Jiayu's work on phage-host interactions has been published in Briefings in Bioinformatics! This study provides a systematic review and benchmark of 27 virus-host prediction tools. By introducing two benchmark datasets—RefSeq-VHDB and MetaHiC-VHDB—the work offers valuable insights into tool performance in distinct scenarios. Congratulations to Jiayu on this important achievement! You can visit here for more information. |
Welcome our new joint Ph.D. student, zewei Li, from the University of Science and Technology of China!Sep. 1, 2025 Zewei Li is a second-year PhD student, under the joint supervision of Professor Yanni Sun and Professor Zhengshuang Hua (USTC). His research focuses on the ecology and evolution of extremophiles. |
We warmly welcome our new Ph.D. students, Shen Yang and Lu Yi!Sep. 1, 2025 We are excited to welcome our new Ph.D. students, Shen Yang and Lu Yi, to the team! Their research interests bring fresh perspectives and expertise that will enrich our ongoing projects. We look forward to their contributions and wish them great success in their academic journey with us! |
Congratulations! Caifang and Jiaojiao's work on dissecting the gut microbial communities and resistomes of wild rats from different ecological areas in Hong Kong was published in Environmental Research!June 7, 2025 Antimicrobial resistance (AMR) is one of the top global public health issues shared across all One Health domains. We trapped 88 live rats, belonging to the species of Rattus norvegicus (n=57), R. tanezumi (n=24), and R. andamanensis (n=7), from city regions, livestock farms, and stables of horse-riding schools (referred to as “suburbs”). We identified 9672 ARGs belonging to 29 ARG types and 554 ARG subtypes. Among them, aminoglycosides, macrolide-lincosamide-streptogramin and chloramphenicol, known to be predominant in livestock gut resistome or manure compost were significantly more abundant in rats from livestock farms. Moreover, some ARGs with high-risk levels, including tetM, tetL, floR, mecR1 and lnuA, as well as plasmid-borne ARGs were significantly more abundant in rats from livestock farms than from city regions or suburbs. Furthermore, zoonotic antimicrobial-resistant bacteria (ARB) were detected, including but not limited to, prioritized antimicrobial-resistant Klebsiella pneumoniae, Proteus mirabilis, Escherichia coli, Enterococcus faecium, Acinetobacter baumannii, Campylobacter jejuni, and Staphylococcus aureus. Notably, resistant zoonotic bacteria of Streptococcus suis, Campylobacter coli, and Campylobacter jejuni were more abundant in wild rats from livestock farms. Our findings provides insights into the gut resistomes and zoonotic bacteria in wild rats in Hong Kong, highlighting the potential role of wild rats in the dissemination of ARGs and zoonotic pathogens, especially for those from agricultural settings. |
Congratulations! Fuchang and jiayu’s work on NCLDV identification from metagenomic data was accepted by ISMB/ECCB 2025Apr. 7, 2025 Nucleocytoplasmic large DNA viruses (NCLDVs) are notable for their large genomes and extensive gene repertoires. However, identifying NCLDV sequences in metagenomic data remains challenging due to their high genomic diversity, limited reference genomes, and shared regions with other microbes. Existing alignment-based and machine learning methods struggle with achieving optimal trade-offs between sensitivity and precision. In this work, we present GiantHunter, a reinforcement learning-based tool for identifying NCLDVs from metagenomic data. By employing a Monte Carlo tree search strategy, GiantHunter dynamically selects representative non-NCLDV sequences as the negative training data, enabling the model to establish a robust decision boundary. Benchmarking on rigorously designed experiments shows that GiantHunter achieves high precision while maintaining competitive sensitivity, improving the F1-score by 10% and reducing computational cost by 90% compared to the second-best method. |
Congratulations! Wei’s work on plasmid multiple host range prediction was published by BioinformaticsFeb. 25, 2025 We present a new plasmid host range prediction tool called MOSTPLAS. To the best of our knowledge, MOSTPLAS is the first attempt to implement a multi-label learning model to recognize all the possible host genera of input plasmid sequences. To mitigate the limitation of lacking databases with complete annotations of broad-host-range plasmids for the training of learning models, we design a plasmid-encoded protein-based pseudo-label generation algorithm and a self-correction asymmetric loss in MOSTPLAS. On the multi-host RefSeq plasmid test set, when all the tools achieved a precision higher than 80%, the recall and F1-score of MOSTPLAS improved by 5.7% and 5.0%, comparing with the second-best tool. In the same time, MOSTPLAS saved about 90% running time compared to BLAST (alignment-based tool) and 80% running time comparing to HOTSPOT (learning-based tool). These results demonstrate the effectiveness and high computation efficiency of MOSTPLAS. You can visit here for more information. |
Congratulations! Jiaojiao’s work on phage proteins’ function prediction was published by BIBJan. 22, 2025 We propose a new protein function annotation tool for phages by leveraging the modular genomic structure of phage genomes. By employing embeddings from the latest protein foundation models and Transformer to capture contextual information between proteins in phage genomes, GOPhage surpasses state-of-the-art methods in annotating diverged proteins and proteins with uncommon functions by 6.78% and 13.05% improvement, respectively. You can visit here for more information. |
Congratulations to Jiayu and Cheng for the updating PhaBOX2!Nov. 10, 2024 We are excited to present PhaBOX2, a state-of-the-art web server designed for comprehensive virus characterization. PhaBOX2 offers a suite of tools for new virus discovery from metagenomic data, quantification of viral contig contamination, virus-host prediction, lifestyle analysis, and taxonomic classification. To the best of our knowledge, PhaBOX2 is among the first web servers to provide such an extensive and user-friendly platform for virus analysis. PhaBOX2 is a significant upgrade to our previously published web server (referred to as PhaBOX hereafter). While the original PhaBOX focused on bacteriophage analysis, PhaBOX2 extends its capabilities to analyze all types of viruses. The server combines alignment-based strategies with state-of-the-art deep learning models, including graph neural networks and large language models, to learn diverse sequence-based features. Notably, all modules have been rigorously validated through peer-reviewed publications, and our research collectively received over 400 citations, demonstrating their significant impact in the field. Key improvements include:1. Extension of analysis capabilities from 10 ICTV classes (6,370 phages) to all virus types, (41 classes and 17,908 viruses). 2. New modules for contamination detection, vOTU grouping, marker gene annotation, and phylogenetic analysis. 3. Significant hardware upgrade enabling 80% faster processing speed. 4. Enhanced user interface with comprehensive documentation. 5. Access to intermediate files and datasets. 6. Local version implementation for large-scale analyses. Since its launch, PhaBOX2 has garnered over 500,000 page views and 10,000 submissions, demonstrating its significant impact in virus research. We maintain a network of 300+ researchers who receive regular updates and provide feedback. Many users have acknowledged the utility of our server, reflecting its value to the virus research community. |
Congratulations! Yongxin’s work on plasmid proteins’ function prediction was accepted for publication by GigaScience.Nov. 27th, 2024 In this study, we introduce PlasGO, a tool that leverages a hierarchical architecture to predict GO terms for plasmid proteins. PlasGO utilizes a powerful protein language model to learn the local context within protein sentences and a BERT model to capture the global context within plasmid sentences. Additionally, PlasGO allows users to control the precision by incorporating a self-attention confidence weighting mechanism. The experimental results collectively demonstrate that PlasGO has achieved commendable performance. PlasGO significantly expanded the annotations of the plasmid-encoded protein database by assigning high-confidence GO terms to over 95% of previously unannotated proteins, showcasing impressive precision of 0.8229, 0.7941, and 0.8870 for the 3 GO categories, respectively, as measured on the novel protein test set. PlasGO is available at HOTSPOT and you can check out the paper here. |
Congratulations to Jiayu for the publication of "Accurate and efficient protein embedding using multi-teacher distillation learning" on Bioinformatics.Sep. 22, 2024 We present a novel protein embedding method using multi-teacher distillation learning to address the computational challenges in protein analysis. Traditional protein embedding methods, while effective for tasks like gene ontology and protein structure prediction, often require massive computational resources with millions to billions of parameters. Our approach leverages knowledge from multiple pre-trained protein embedding models to create efficient protein representations. The method demonstrates significant efficiency improvements, reducing computational time by approximately 70% while maintaining accuracy within ±1.5% of state-of-the-art models. This advancement enables more efficient large-scale protein analysis in bioinformatics research. |
Congratulations to Yilin and GuoWei for the publication of "VirTAXA: enhancing RNA virus taxonomic classification with remote homology search and tree-based validation" on Bioinformatics.Sep. 22, 2024 We present VirTAXA, a robust classification tool that combines remote homology search and tree-based validation to enhance the genus-level taxonomic classification of RNA viruses. VirTAXA can predict the genus label of an assembled viral contig and provide evidence type for each prediction. It achieves comparable accuracy to state-of-the-art methods while assigning genus labels to a greater number of sequences. Specifically, on the Global Ocean RNA metatranscriptomic data, VirTAXA can assign genus labels for over 10% more contigs than other classification tools. Furthermore, we demonstrated that VirTAXA can be conveniently extended to other types of viruses. VirTAXA takes contigs as input, assigns the genus label, and provides the associated evidence. It can also predict new genera using the phylogenetic tree analysis. More information can be found in the paper "VirTAXA: enhancing RNA virus taxonomic classification with remote homology search and tree-based validation". |
Congratulations to Xubo for his outstanding work in developing a new tool named SegVir for constructing segmented viruses!Aug. 10, 2024 The article titled "SegVir: Reconstruction of Complete Segmented RNA Viral Genomes from Metatranscriptomes" has been accepted by Molecular Biology and Evolution. Our tool, "SegVir" can identify viral segments, reconstruct full genomes, and quantify the completeness of segmented viruses. This is a collaborative effort with Prof. Shi Mang, who provided invaluable inspiration and suggestions throughout the project. |
Congratulations to Guowei on his paper "RNAVirHost: A Machine Learning-Based Method for Predicting Hosts of RNA Viruses Through Viral Genomes" on RNA virus-host identification accepted by GigaScience!Aug. 10, 2024 In collaboration with Dr. Jiang Jinzhe, our initial goal was to identify hosts of ocean viruses, a highly challenging problem due to the diversity and complexity of potential hosts. Guowei excelled in formulating a feasible computational problem for practical host prediction. There remains a significant need for better host prediction solutions. In our discussion section, we have summarized and visualized the major challenges. |
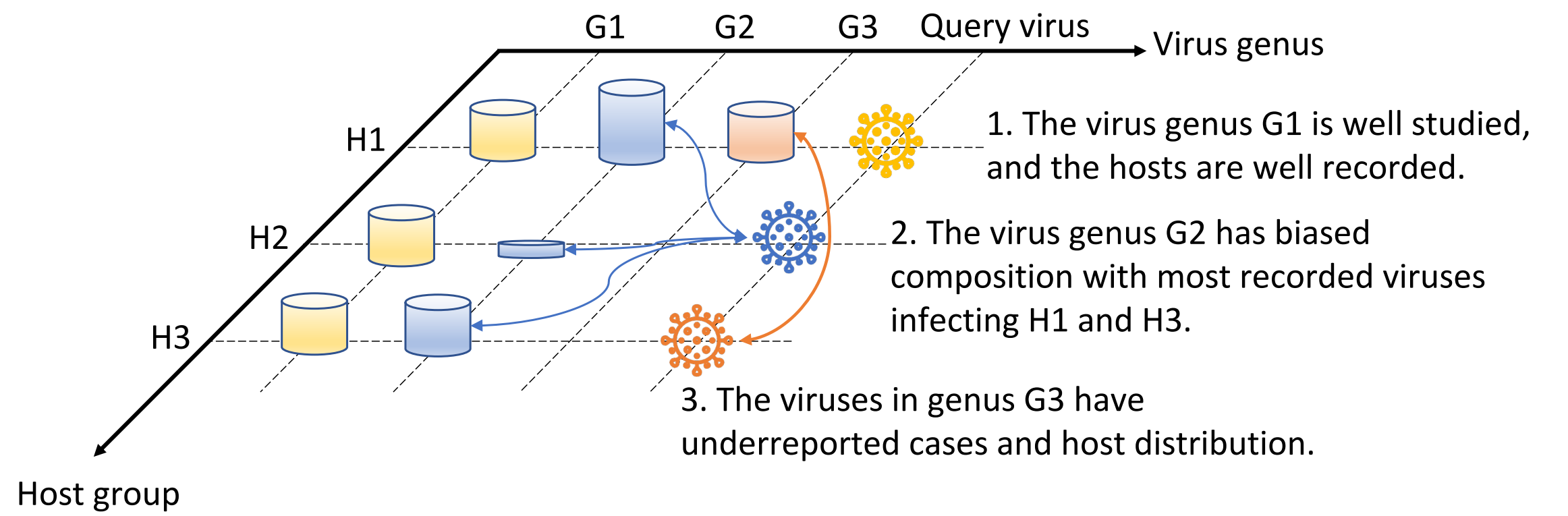
A visualization illustrating the potential sequencing bias in the reference database, posing a challenge to the prediction of hosts. On the virus genus axis, there are three reference genera represented by colors: yellow for G1, blue for G2, and red for G3. The host group axis consists of three distinct host groups (H1, H2, H3) present in the reference database. The cylinders at the intersections of the dashed lines represent viruses belonging to the respective genera that infect the corresponding host groups. The height of each cylinder indicates the relative number of viruses. G1 is extensively studied, and its hosts are well-documented. G2 and G3 are under-studied, resulting in limited information about their hosts. More information can be found in the paper: “RNAVirHost: A Machine Learning-Based Method for Predicting Hosts of RNA Viruses Through Viral Genomes,” which will appear in GigaScience soon. |
Congratulations to Dr. Yu Runzhou and Ziyi on publishing a review of using profile hidden Markov models (pHMMs) for virus discovery in metagenomic data!Aug. 10, 2024 We conducted a thorough comparison and evaluation of multiple pHMM databases, including comprehensive ones like Pfam and virus-specific databases like CheckV. While using pHMM for RNA virus search is generally effective, finding DNA viruses (such as phages) remains challenging with these databases. Some virus-specific models incur a high false positive rate. More information can be found in the paper: “Utilizing Profile HMM Databases for Discovering Viruses from Metagenomic Data: A Comprehensive Review,” available at Briefings in Bioinformatics: https://pubmed.ncbi.nlm.nih.gov/39003531/. |
Congratulations! Ziyi and Dehan's work on microbial source tracking has been accepted for presentation at ISMB 2024 and will be included in the conference proceedings, which will be published online in the journal Bioinformatics.April 11, 2024 This work developed a novel tool called SourceID-NMF for more precise microbial source tracking. SourceID-NMF utilizes a non-negative matrix factorization (NMF) algorithm to trace the microbial sources contributing to a target sample. By leveraging the taxa abundance in both available sources and the target sample, SourceID-NMF estimates the proportion of available sources present in the target sample. A series of benchmarking experiments using simulated and real data were conducted to evaluate the performance of SourceID-NMF. The simulated experiments aimed to mimic realistic yet challenging scenarios, including identifying highly similar sources, irrelevant sources, unknown sources, low abundance sources, and noise sources. The results demonstrate the superior accuracy of SourceID-NMF compared to existing methods. In particular, SourceID-NMF accurately estimated the proportion of irrelevant and unknown sources, while other tools tended to either over- or under-estimate them. Additionally, the noise sources experiment showcased the robustness of SourceID-NMF for microbial source tracking. SourceID-NMF is available online at <a href= https://github.com/ZiyiHuang0708/SourceID-NMF> here. |
Congratulations! Herui has been accepted into the MIT-Novo Nordisk AI Postdoc Program.Feb. 9, 2024 The MIT-Novo Nordisk Artificial Intelligence Postdoctoral Fellows Program supports postdoctoral fellows conducting research at the intersection of AI and data science with life sciences. Each year, the program will support a cohort of up to ten postdoctoral fellows for two-year terms. Postdoctoral fellows participating in the program will receive professional development opportunities, including entrepreneurship-focused workshops and mentorship from experts in both life science and data science. You can visit this website for more information. |

|
Congratulations! Herui and Runzhou Successfully Defend Their Dissertations!The thesis title of Herui is "Computational Methods for high-resolution microbial composition analysis and relevant applications". In this thesis, we developed a viral strain identification tool VirStrain, a bacterial strain identification tool StrainScan, and a microbial-based host disease classification method GDmicro. We applied these tools to large-scale real sequencing samples, including pathogen-infected patients, individuals from different countries, colorectal cancer cohorts, etc. The experimental results demonstrate these tools have the potential to enhance our comprehension of microbes and facilitate the development of diagnostic and therapeutic strategies for diseases. Runzhou's thesis title is "Computational Methods for accurate reconstruction of RNA viral genomes using third-generation sequencing data". This thesis presents three main works. The first work introduces AccuVIR, a tool for accurate viral genome assembly and polishing by generating high-quality paths in alignment graphs and then ranking them using multiple criteria. It performs well with both known and novel viruses, as demonstrated through evaluations with simulated and real sequencing data. The second work is HMMPolish, a pipeline designed to refine protein-coding regions of known RNA viruses sequenced using third-generation sequencing. HMMPolish utilizes profile Hidden Markov Models (pHMMs) of viral proteins and has been validated with clinically important virus datasets. The third work reviews commonly used profile HMM databases for viral metagenomic data. It evaluates their model properties using quantitative metrics and assesses their performance with simulated and real metagenomic datasets. The findings provide practical suggestions for optimizing the use of these databases in viral metagenomics research. |
Congratulations! Herui and Jiayu's work GDmicro was accepted for publication by Bioinformatics.Dec. 12th, 2023 With advances in metagenomic sequencing technologies, there are accumulating studies revealing the associations between the human gut microbiome and some human diseases. These associations shed light on using gut microbiome data to distinguish case and control samples of a specific disease, which is also called host disease status classification. However, available tools have not fully addressed two challenges associated with this task: limited labeled microbiome data and decreased accuracy in cross-studies. To address these challenges, we develop a new tool GDmicro, which combines semi-supervised learning and domain adaptation to achieve a more generalized model using limited labeled samples. GDmicro is available at GDmicro Check out the paper at GDmicro: classifying host disease status with GCN and deep adaptation network based on the human gut microbiome data. |
Congratulations! Jiaojiao and Cheng's work PhaGenus was Published on the BIB.Nov. 15, 2023 Bacteriophages prey on and replicate within bacterial cells and have a significant role in modulating microbial communities. However, the taxonomic classification of assembled phage contigs still faces several challenges. In this work, we develop a learning-based model named PhaGenus, which conducts genus-level taxonomic classification for phage contigs. PhaGenus utilizes a powerful Transformer model to learn the association between protein clusters and support the classification of up to 508 genera. For more information refer to the paper. PhaGenus: genus-level classification of bacteriophages using a Transformer model. |
Congratulations! Our cooperation works with Chinese Academy of Sciences "Identifying ARG-carrying bacteriophages in a lake replenished by reclaimed water using deep learning techniques" was accepted for publication by Water Research.Nov. 10, 2023 As important mobile genetic elements, phages support the spread of antibiotic resistance genes (ARGs). Previous analyses of metaviromes or metagenome-assembled genomes (MAGs) failed to assess the extent of ARGs transferred by phages, particularly in the generation of antibiotic pathogens. Therefore, we have developed a bioinformatic pipeline that utilizes deep learning techniques to identify ARG-carrying phages and predict their hosts, with a special focus on pathogens. Using this method, we discovered that the predominant types of ARGs carried by temperate phages in a typical landscape lake, which is fully replenished by reclaimed water, were related to multidrug resistance and β-lactam antibiotics. Check out the paper at Identifying ARG-carrying bacteriophages in a lake replenished by reclaimed water using deep learning techniques. |
Congratulations! Jiayu was the CHAMPION in the competition named “Postgraduate Student Research Paper Competition 2022 – 2023”.Aug. 26, 2023 The competition is held by IEEE (Hong Kong) Computational Intelligence Chapter. Jaiyu was the CHAMPION for his outstanding academic performance. |

|
Congratulations! Herui and Yongxin’s work for identifying bacterial strains from short reads was accepted for publication by Microbiome.Aug. 17th, 2023 In this work, we developed a novel software, StrainScan, for identifying bacterial strains from short read. By utilizing a novel tree-based k-mers indexing structure, StrainScan can strike a balance between the strain identification accuracy and the computational complexity. We tested StrainScan extensively on a large number of simulated and real sequencing data and benchmarked StrainScan with popular strain-level analysis tools. The results show that StrainScan has higher accuracy and resolution than the state-of-the-art tools on strain-level composition analysis. It improves the F1 score by 20% in identifying multiple strains at the strain level. StrainScan is available at StrainScan. Check out the paper at High-resolution strain-level microbiome composition analysis from short reads. |
We participated in the ISMB/ECCB 2023 conference!Aug. 02, 2023 Congratulations! Dr. Sun and four talented Ph.D. students, namely Jiayu Shang, Xubo Tang, Dehan Cai, and Cheng Peng, recently participated in the prestigious ISMB/ECCB 2023 conference from 23rd to 27th July. During the conference, Jiayu Shang delivered an outstanding proceedings presentation titled PhaVIP: Phage VIrion Protein Classification based on chaos game representation and Vision Transformer. Xubo Tang showcased his work on identifying plasmid contigs from metagenomic data using a Transformer , while Dehan Cai presented his report on HaploDMF: viral haplotype reconstruction from long reads via deep matrix factorization. Cheng Peng exhibited the work on PhaBOX: A web server for identifying and characterizing bacteriophage contigs in metagenomic data through a poster presentation. Their innovative research work has garnered significant attention from the scientific community. If you are interested in learning more about their work, please refer to the published papers. You can visit this website for more information. |

|
Congratulations! Runzhou and Umer’s work for correcting (polishing) errors in protein-coding regions of RNA viruses was accepted for publication by Briefings in Bioinformatics.July 21th, 2023 In this work, we developed a novel pipeline, HMMPolish, for correcting (polishing) errors in protein-coding regions of known RNA viruses. By utilizing profile Hidden Markov Models of protein families/domains in known viruses, HMMPolish can correct errors that are ignored by available polishers. We extensively validated HMMPolish on 34 datasets that covered four clinically important viruses, including HIV-1, influenza-A, norovirus, and severe acute respiratory syndrome coronavirus 2. These datasets contain reads with different properties, such as sequencing depth and platforms (PacBio or Nanopore). The benchmark results against popular/representative polishers show that HMMPolish competes favorably on error correction in coding regions of known RNA viruses. HMMPolish is available at HMMPolish . Check out the paper at HMMPolish: a coding region polishing tool for TGS-sequenced RNA viruses. |
Congratulations! Xubo, Jiayu and Yongxin’s work on plasmid identification was accepted for publication by Nucleic Acids Research.Jun. 24, 2023 Plasmids are mobile genetic elements that carry crucial accessory genes. Cataloging plasmids is a fundamental step in elucidating their role in promoting horizontal gene transfer between bacteria. To identify plasmid contigs from short-read assemblies, we developed a tool called PLASMe that utilizes the Transformer. PLASMe leverages the strengths of both alignment and learning-based methods. The alignment component in PLASMe facilitates the easy identification of closely related plasmids, while order-specific Transformer models predict diverged plasmids with accuracy. The tool is available at PLASMe. Check out the paper at PLASMe: a tool to identify PLASMid contigs from short-read assemblies using transformer |
Congratulations! Xubo received 2nd Prize for the presentation titled “Identify Plasmid from Metagenomic Data Using Transformer” in the EE Graduate Research Seminar Awards.May 10, 2023 |
Congratulations! Yongxin, Jiayu, and Xubo’s work on plasmid host prediction was accepted for publication by Bioinformatics!Apr. 19, 2023 In this work, we construct a tool named HOTSPOT, aiming at predicting the host association of plasmids. By incorporating the state-of-the-art language model, Transformer, in each node’s taxon classifier, the top-down tree search achieves an accurate host taxonomy prediction for the input plasmid contigs. We rigorously tested HOTSPOT on multiple datasets and all experiments show that HOTSPOT outperforms other popular methods. The tool is available at HOTSPOT Check out the paper at HOTSPOT: hierarchical host prediction for assembled plasmid contigs with transformer |
Congratulations! The Chinese version of VirBot is published at the offical wechat account "宏基因组"Apr. 16, 2023 You can visit detailed information here. |
Congratulations! Guowei and Xubo’s work on RNA virus identification and taxonomy was accepted for publication by Bioinformatics.Feb. 17, 2023 In this work, we described a tool, VirBot, aiming at identifying the RNA virus contig from the metagenomic data. Based on the profile hidden Markov model (pHMM), VirBot can better detect novel RNA virus sequences which share little similarity with the known species while maintaining high precision. In the benchmark experiment on both simulated and real sequencing data, VirBot shows its high specificity in metagenomic datasets and superior sensitivity in detecting novel RNA viruses. The tool is available at VirBot.Check out the paper at VirBot: an RNA viral contig detector for metagenomic data |
A new integrated software named PhaBOX for phage identification and analysis has been developed!Jan. 3rd, 2023 Bacteriophages (phages) play key roles in regulating the composition/function of the microbiome by infecting their host bacteria. Lacking integrated software for phage identification and analysis, novel phages awaiting to be discovered constitute a large portion of “viral dark matter”. In this work, we developed a web server, named PhaBOX, to accurately identify and analyze phage contigs in metagenomic data. To our best knowledge, this is the first web server for comprehensive phage contig analysis in metagenomic data. PhaBOX integrates our previously published tools: PhaMer, PhaTYP, PhaGCN, and CHERRY, for phage identification, lifestyle prediction, taxonomy classification, and host prediction, respectively. To help users conduct downstream analysis, PhaBOX also provides visualization of the essential features for making the predictions, such as the similarity-based relationships between the contigs and other phages, predicted proteins on the contigs, and protein homology. All the predictions and intermediate results are provided for users. We hope that it can help advance the field of phage study in various ecosystems. To try PhaBOX, please click the link PhaBOX |
Congratulations! Runzhou and Dehan’s work on viral genome assembly and polishing was accepted for publication by Bioinformatics.Dec. 26th, 2022 In this work, we introduce a new tool, AccuVIR, for viral genome assembly and polishing using error-prone long reads. It can better distinguish sequencing errors from true variants based on the key observation that sequencing errors can disrupt the gene structures of viruses, which usually have high density of coding regions. Our experimental results on both simulated and real third-generation sequencing data demonstrated its superior performance on generating more accurate viral genomes than generic assembly or polish tools. The tool is available at https://github.com/rainyrubyzhou/AccuVIR. Check out the paper at https://doi.org/10.1093/bioinformatics/btac827 |
Congratulations! Yilin, Jiayu and Peng Cheng's work on phage taxonomy classification was accepted for publication by Frontiers in Microbiology.Dec. 22rd, 2022 This is the first review conducted under the new ICTV classification framework since several large families were removed from ICTV in August 2022. This study provides a comprehensive review of phage classification in different scenarios and a practical guidance for choosing appropriate taxonomic classification pipelines. (https://www.frontiersin.org/articles/10.3389/fmicb.2022.1032186) |
Congratulations! Dehan and Jiayu’s work on viral haplotype reconstruction was accepted for publication by Bioinformatics.Oct. 21th, 2022 This work developed a tool “HaploDMF” to reconstruct viral haplotypes from TGS data. Unlike existing tools that reconstruct haplotypes by checking the identity of overlap between reads, HaploDMF utilizes a deep matrix factorization model with an adapted loss function to automatically learn latent features from aligned reads. It is able to achieve highly robust performance on data with different properties while existing tools’ performance can be affected by the overlap size between reads. The tool is available at https://github.com/dhcai21/HaploDMF. |
Congratulations! Jiayu's work on phage lifestyle prediction is accepted today in Briefings in Bioinformatics!Oct. 17th, 2022 Bacteriophages (or phages), which infect bacteria, have two distinct lifestyles: virulent and temperate. Predicting the lifestyle of phages helps decipher their interactions with their bacterial hosts, aiding phages' applications in fields such as phage therapy. PhaTYP adopt Bidirectional Encoder Representations from Transformer (BERT) to learn the protein composition and associations from phage genomes and achieves more stable performance on short contigs. arXiv version: [PhaTYP: Predicting the lifestyle for bacteriophages using BERT ](https://arxiv.org/abs/2206.09693) |
Congratulations! Jiayu, Herui, Xubo, and Dehan received awards!Sep. 9, 2022 They got the Research Tuition Scholarship ($42,096) and the Outstanding Academic Performance Award ($1,000) for their outstanding academic performance in the 2021-2022 academic year! |
Congratulations! Jiayu's work on identifying phage is published today in Briefings in Bioinformatics!June 30, 2022 Accurate identification of bacteriophages from metagenomic data using Transformer |
Congratulations! Herui and Dehan received the 2nd Prize in competition of "The 8th Hong Kong University Student Innovation and Entrepreneurship Competition"June 07, 2022 you can vist this website [https://www.hkchallengeplus.com/news/] for more information. |
Herui and Dehan attended the final round of the competition named "The 8th Hong Kong University Student Innovation and Entrepreneurship Competition" at Science Park of Hong KongMay 27, 2022 We recorded a video about our work in the competition. If you are interested in it, please scan this 2D code. |

|

|
Congratulations! Jiayu's work on host prediction is published today in Briefings in Bioinformatics!May 22, 2022 |
Congratulations! Xubo's poster presentation receives the $2,000 for 2nd Prize in EE faculty!May 19, 2022 |
Ph.D student Tang Xubo gave a talk titled "Sensitive RNA Virus Read Binning Using Learning-Based Models"April 27, 2022 The talk was held at a company called Centre for Intelligent Multidimensional Data Analysis Limited |
Welcome!A new research assistant Liyan joined the group todayApril 13, 2022 Welcome Liyan! |
An invited talk: Charactering viral haplotypes using long readsApril 6, 2022 Yanni Sun will give an invited talk “Charactering viral haplotypes using long reads” at the Workshop on Combinatorial Problems of Strings and Graphs and Their Applications in Bioinformatics. This is organized by National University of Singapore and Institute for Mathematical Sciences. Combinatorial Problems of Strings and Graphs and Their Applications in Bioinformatics Part 2 |
Talk: Understanding the language of life using AIMar. 18, 2022 Yanni Sun gave a talk titled “Understanding the language of life using AI” to middle high-school students in Hong Kong at CityU-Learning Classroom. |
Dehan’s work is publishedFeb. 14, 2022 Dehan’s work on viral haplotype reconstruction “Reconstructing viral haplotypes using long reads” is published today! |
Xubo and Jiayu’s work on RNA virus classification is publishedFeb. 7, 2022 "RdRp-based sensitive taxonomic classification of RNA viruses for metagenomic data" is published today! RdRp-based sensitive taxonomic classification of RNA viruses for metagenomic data |
Herui’s work on "strain-level" RNA virus composition analysis is publishedJan. 31, 2022 "VirStrain: a strain identification tool for RNA viruses" is published! |