Research Area
An overview of my recent research interests.
Virus classification using deep learning
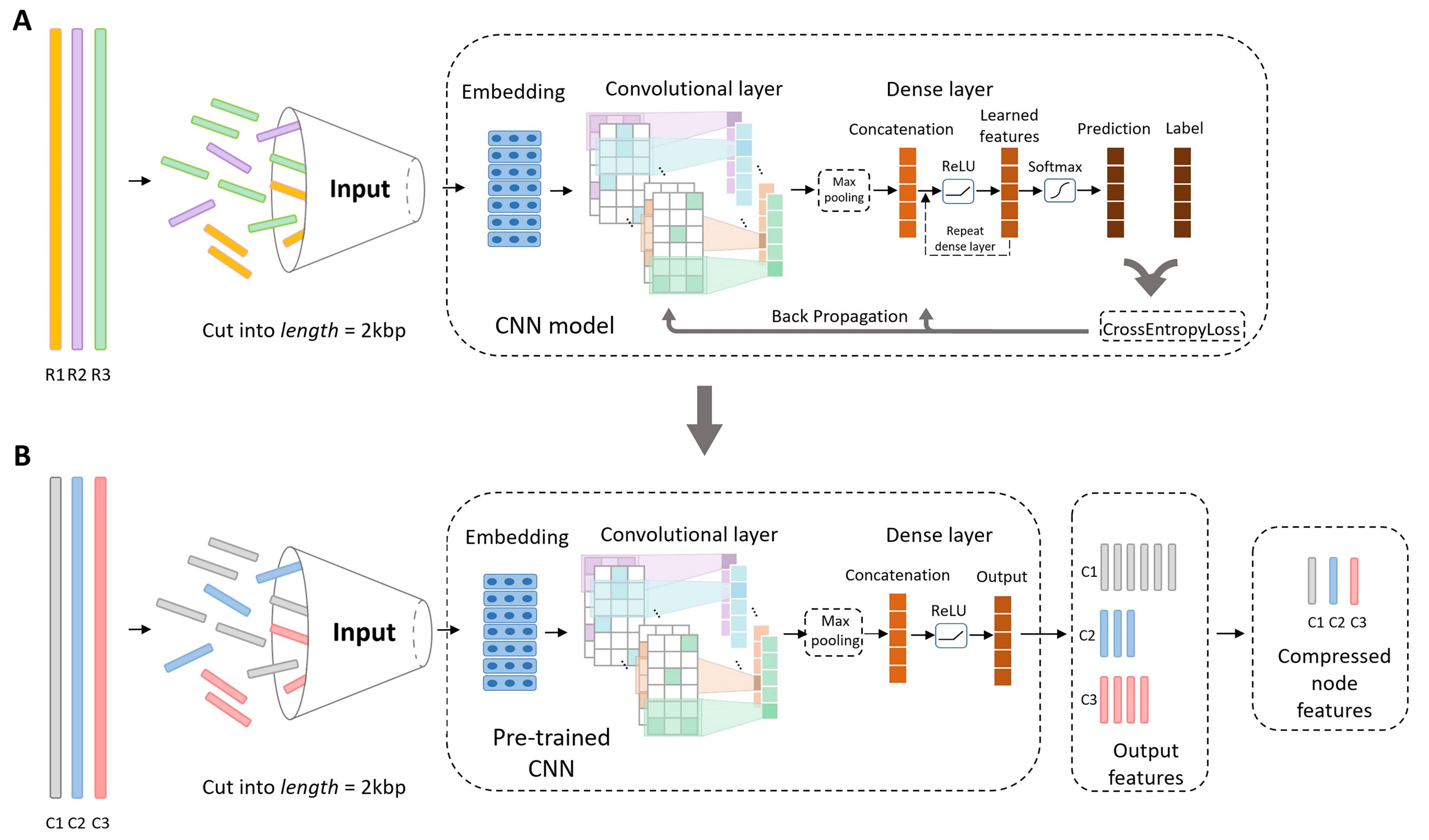
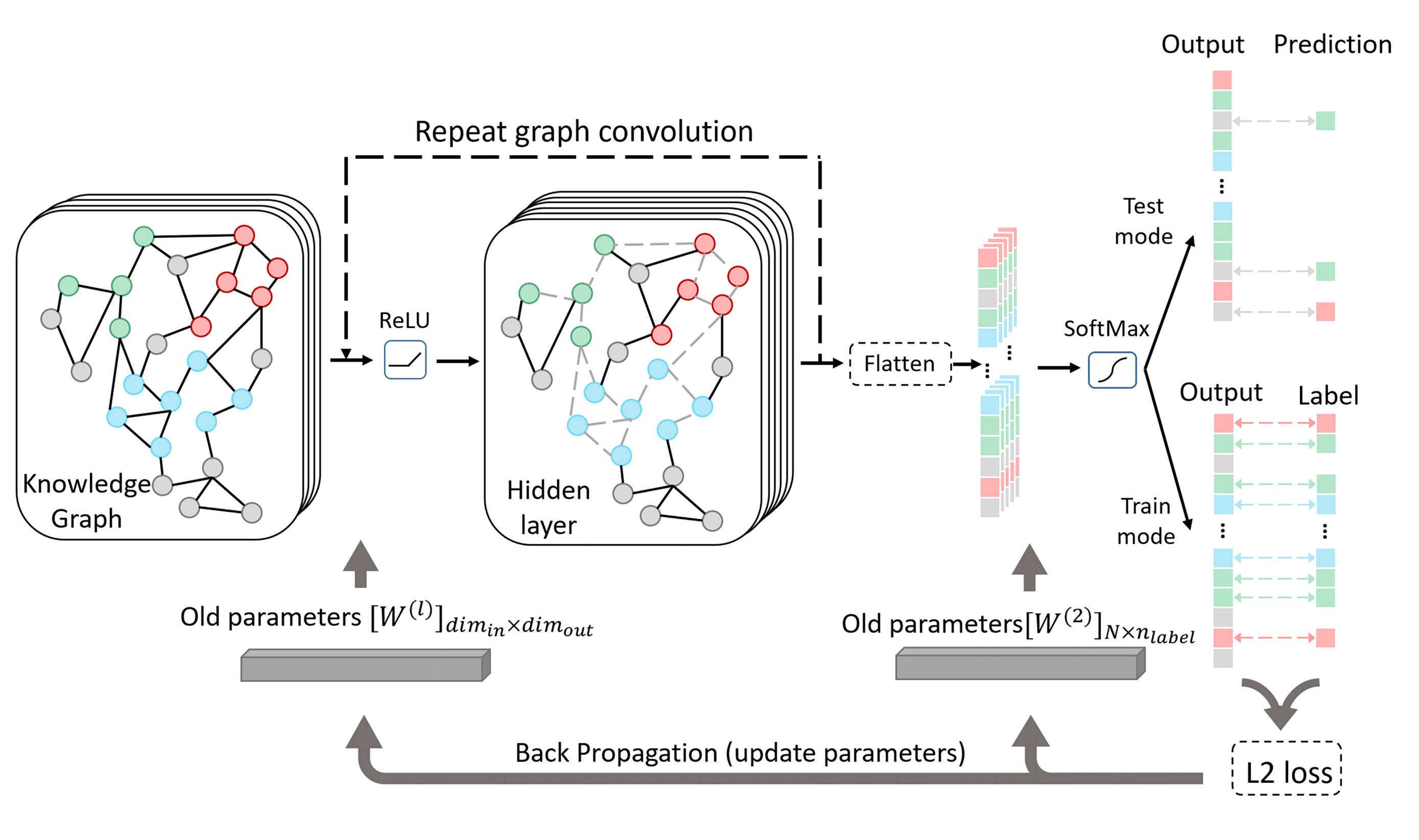
Bacteriophages (aka phages), which mainly infect bacteria, play key roles in the biology of microbes. As the most abundant biological entities on the planet, the number of discovered phages is only the tip of the iceberg. Recently, many new phages have been revealed using high throughput sequencing, particularly metagenomic sequencing. Compared to the fast accumulation of phage-like sequences, there is a serious lag in taxonomic classification of phages. High diversity, abundance, and limited known phages pose great challenges for taxonomic analysis. In particular, alignment-based tools have difficulty in classifying fast accumulating contigs assembled from metagenomic data. We recently developed a novel semi-supervised learning model, named PhaGCN, to conduct taxonomic classification for phage contigs. In this learning model, we construct a knowledge graph by combining the DNA sequence features learned by convolutional neural network (CNN) and protein sequence similarity gained from gene-sharing network. Then we apply graph convolutional network (GCN) to utilize both the labeled and unlabeled samples in training to enhance the learning ability. We tested PhaGCN on both simulated and real sequencing data. The results clearly show that our method competes favorably against available phage classification tools. The source code of PhaGCN is available at https://github.com/KennthShang/PhaGCN
|
|
Virus host prediction
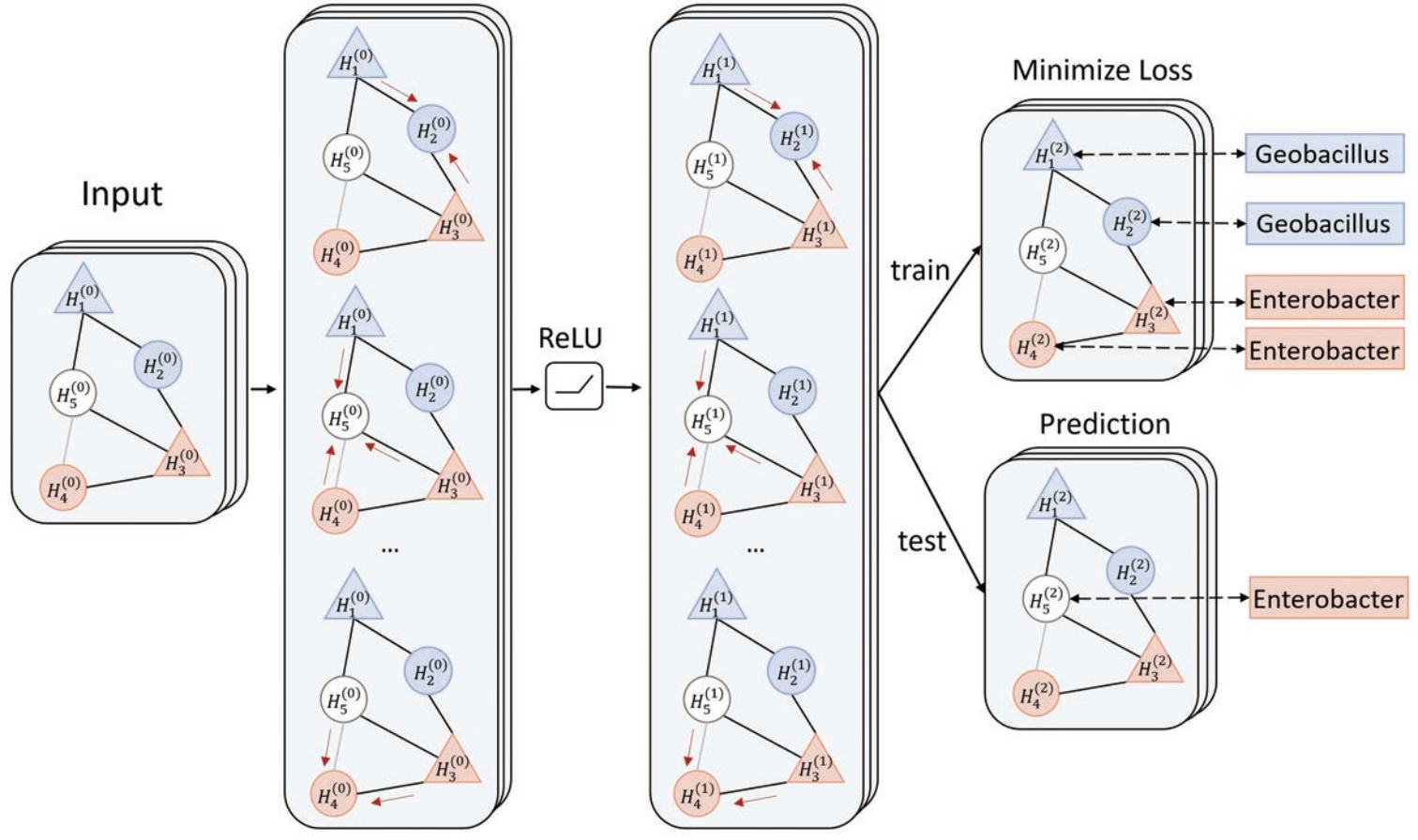
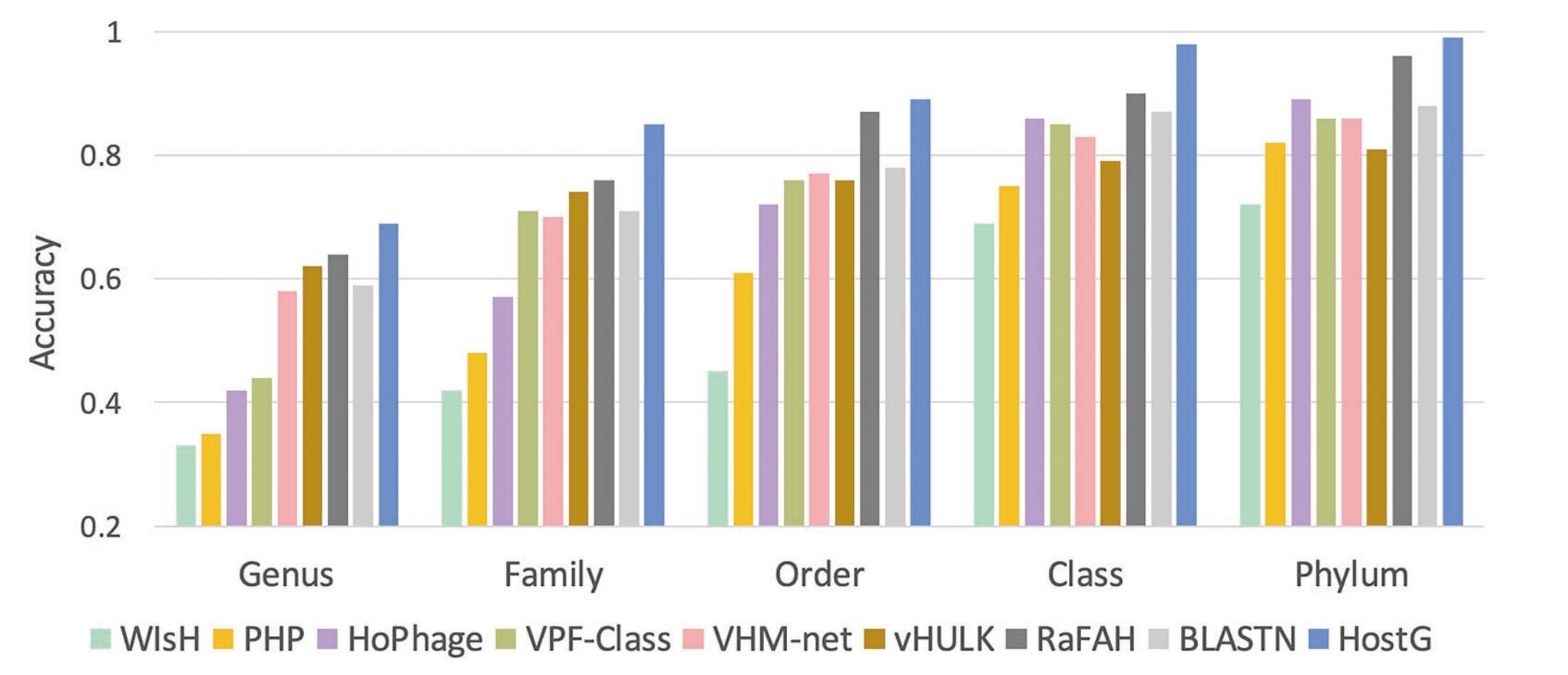
Prokaryotic viruses, which infect bacteria and archaea, are the most abundant and diverse biological entities in the biosphere. To understand their regulatory roles in various ecosystems and to harness the potential of bacteriophages for use in therapy, more knowledge of viral-host relationships is required. High-throughput sequencing and its application to the microbiome have offered new opportunities for computational approaches for predicting which hosts particular viruses can infect. However, there are two main challenges for computational host prediction. First, the empirically known virus-host relationships are very limited. Second, although sequence similarity between viruses and their prokaryote hosts have been used as a major feature for host prediction, the alignment is either missing or ambiguous in many cases. Thus, there is still a need to improve the accuracy of host prediction. We designed a semi-supervised learning model, named HostG, to conduct host prediction for novel viruses. We construct a knowledge graph by utilizing both virus-virus protein similarity and virus-host DNA sequence similarity. Then graph convolutional network (GCN) is adopted to exploit viruses with or without known hosts in training to enhance the learning ability. During the GCN training, we minimize the expected calibrated error (ECE) to ensure the confidence of the predictions. We tested HostG on both simulated and real sequencing data and compared its performance with other state-of-the-art methods specifcally designed for virus host classification (VHM-net, WIsH, PHP, HoPhage, RaFAH, vHULK, and VPF-Class). HostG outperforms other popular methods, demonstrating the efficacy of using a GCN-based semi-supervised learning approach. A particular advantage of HostG is its ability to predict hosts from new taxa. The source code of HostG \cite{HostG} is available at: https://github.com/KennthShang/HostG.
|
|
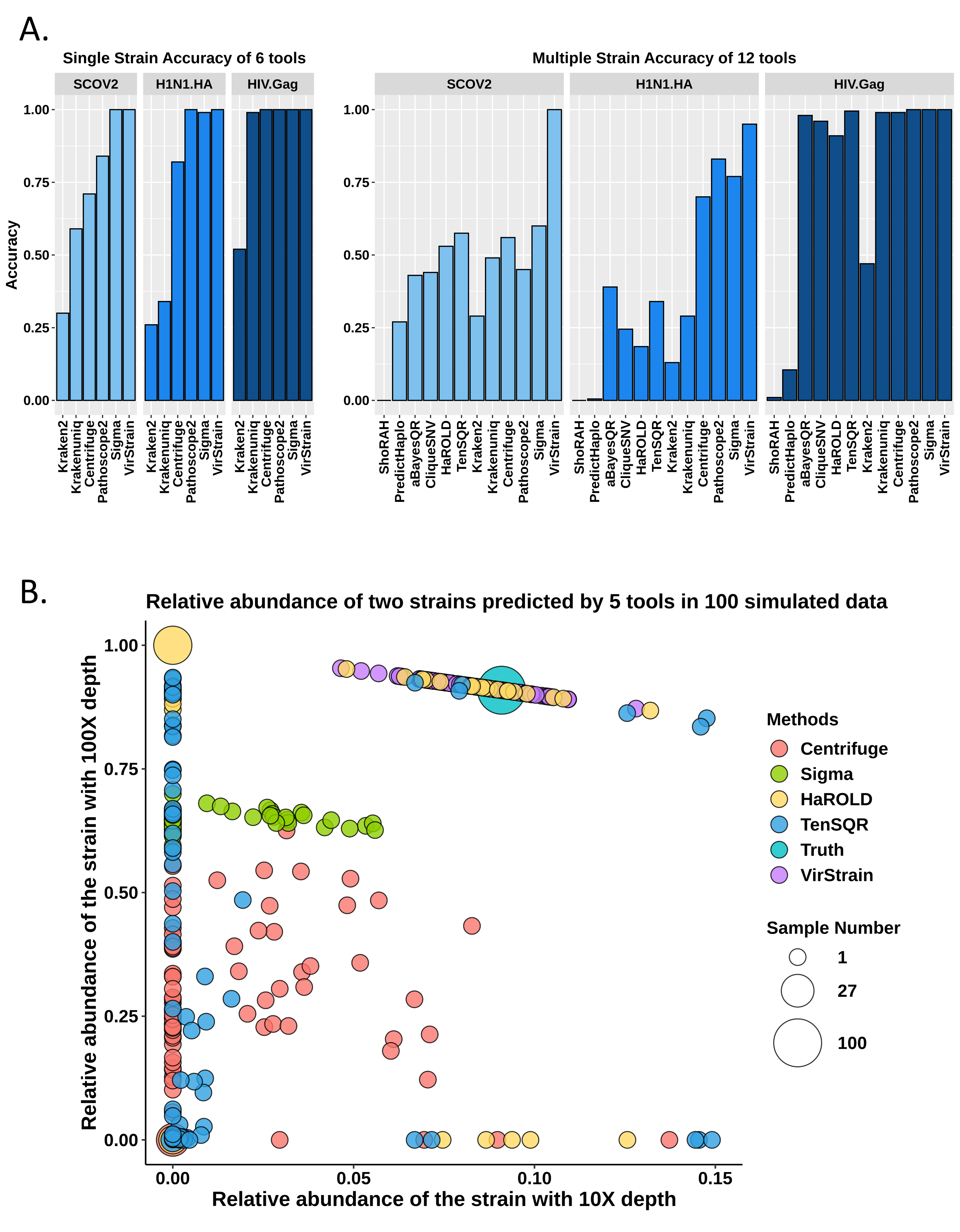
High resolution viral variant identification from short reads
Viruses change constantly during replication, leading to high intra-species diversity. Although many changes are neutral or deleterious, some can confer on the virus different biological properties such as better adaptability. In addition, viral genotypes often have associated metadata, such as host residence, which can help with inferring viral transmission during pandemics. Thus, subspecies analysis can provide important insights into virus characterization. Here, we present VirStrain, a tool taking short reads as input with viral strain composition as output. We rigorously test VirStrain on multiple simulated and real virus sequencing datasets. VirStrain outperforms the state-of-the-art tools in both sensitivity and accuracy. The source code of VirStrain is freely available at https://github.com/liaoherui/VirStrain.
|
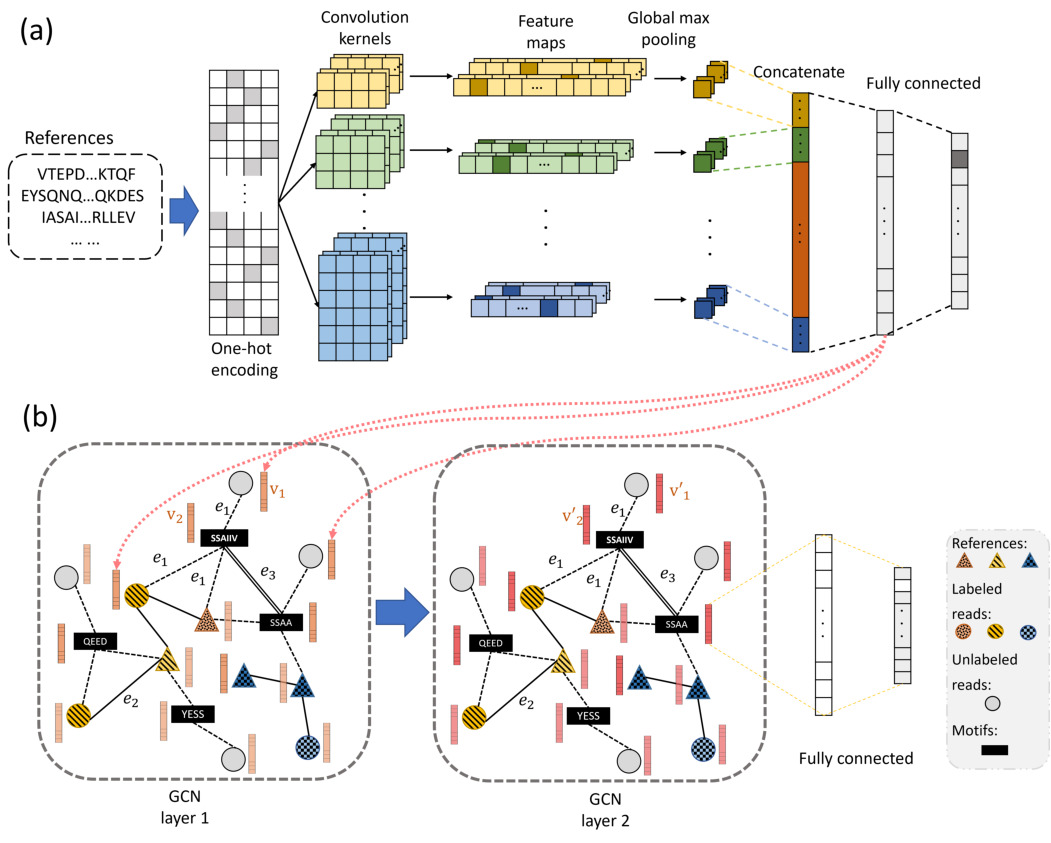
Read-level RNA virus identification
With advances in library construction protocols and next-generation sequencing technologies, viral metagenomic sequencing has become the major source for novel virus discovery. Conducting taxonomic classification for metagenomic data is an important means to characterize the viral composition in the underlying samples. However, RNA viruses are abundant and highly diverse, jeopardizing the sensitivity of comparison-based classification methods. To improve the sensitivity of read-level taxonomic classification, we developed a RdRp gene-based read classification tool RdRpBin. It combines alignment-based strategy with machine learning models in order to fully exploit the sequence properties of RdRp. We tested our method and compared its performance with the state-of-the-art tools on the simulated and real sequencing data. RdRpBin competes favorably with all. In particular, when the query RNA viruses share low sequence similarity with the known viruses (~0.4), our tool can still maintain a higher F-score than the state-of-the-art tools. The experimental results on real data also showed that RdRpBin can classify more RNA viral reads with a relatively low false-positive rate. Our tool is implemented in Python and can be downloaded at https://github.com/HubertTang/RdRpBin.
|
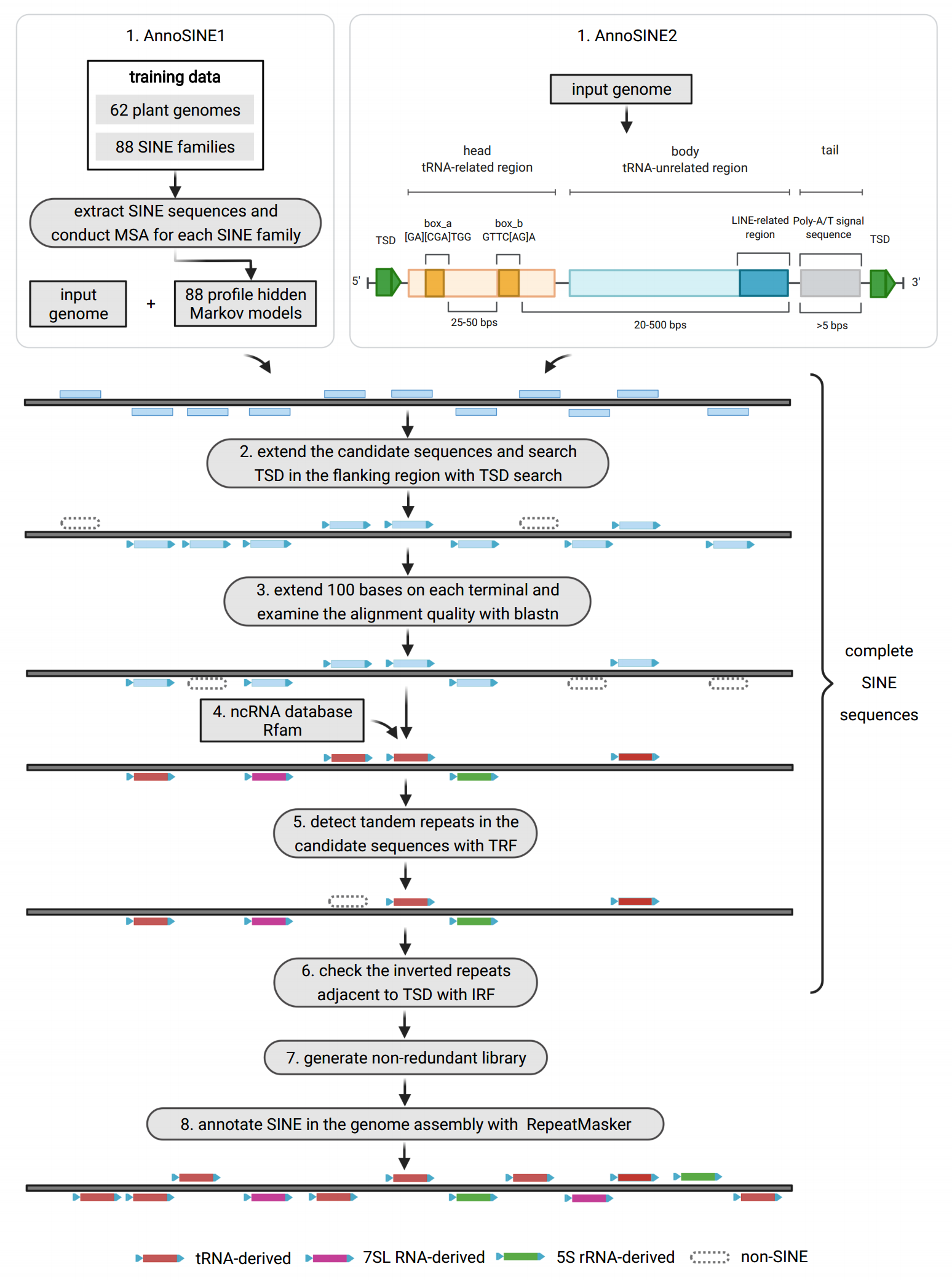
Annotating Short interspersed nuclear elements (SINEs) in plant genomes
Short interspersed nuclear elements (SINEs) are a widespread type of small transposable element (TEs). With increasing evidence for their impact on gene function and genome evolution in plants, accurate genome-scale SINE annotation becomes a fundamental step for studying the regulatory roles of SINEs and their relationship with other components in the genomes. Despite overall promising progress made on TE annotation, SINE annotation remains a major challenge. Unlike some other TEs, SINEs are short and heterogeneous; and they usually lack well-conserved sequence or structural features. Thus, current SINE annotation tools have either low sensitivity or high false discovery rates. Given the demand and challenges, we made an attempt to provide an accurate and efficient SINE annotation tool for plant genomes. The pipeline starts with maximizing pool of SINE candidates via profile hidden Markov model (pHMM)-based homology search and \textit{de novo} SINE search using structural features. Then, it excludes the false positives by integrating all known features of SINEs and the features of other types of TEs that can be often misannotated as SINEs. As a result, the pipeline significantly improves the tradeoff between sensitivity and accuracy, with both values close to or over 90\%. We tested our tool in \at\ and \os, and the results show that our tool competes favorably against existing SINE annotation tools. The simplicity and effectiveness of this tool would be potentially useful for generating more accurate SINE annotations for other plant species. The pipeline is freely available at https://github.com/yangli557/AnnoSINE.
|
Other projects’ topics can be found in the following figure.

|